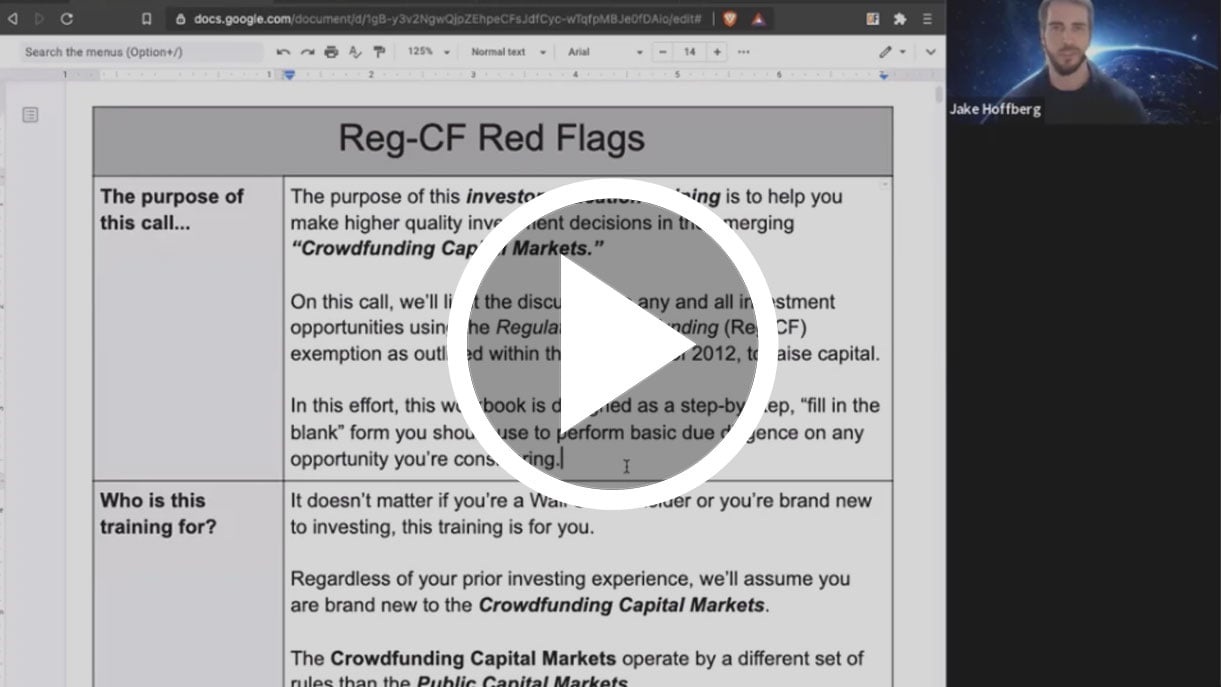
If you’re looking to get in on the exciting world of breakthrough medical devices, there’s one thing you need to know about these new technologies before you invest a single penny…
The FDA medical device approval process.
It doesn’t matter how slick the marketing material is, if the management team can’t navigate this process and bring their product to market, that’s it.
Or as the late Bill Paxton once said in the movie Aliens, “Game over man, game over!”
Today, we’ll walk you through the entire process so you know how it works (and what questions to ask management if you find a deal you like).
A Brief Primer on Medical Devices
Now, before we get too far into the weeds and I lose you — or you swear to never invest in MedTech because it sounds too complex — the main things you really need to know before investing are…
- How is this device classified?
- Has it been granted 510(k) clearance?
- Does it require a Pre-Market Approval?
- And, what are the Post-Market Studies requirements?
Answer those and you’ll have a great starting point to better analyze a potential investment.
But let’s dig in further. First thing’s first, let’s get some definitions out of the way…
According to Section 201(h) of the Federal Food, Drug, and Cosmetic Act, a medical device is:
“any instrument, machine, contrivance, implant, in vitro reagent that’s intended to treat, cure, prevent, mitigate, or diagnose disease in man.”
Some examples could be a simple tongue depressor, or a thermometer, all the way to an advanced robotic surgical device.
These devices are regulated by the Food and Drug Administration (FDA) – and more specifically, a division inside the FDA called Center for Devices and Radiological Health (CDRH) – who are expected to guarantee the safety and effectiveness of these devices.
[Note: the FDA was given the authority to begin regulating all medical devices on May 28, 1976.]
Inside the CDRH are six offices that all serve different purposes…
- Office of Compliance – responsible for ensuring that all medical devices, once placed on the market, remain safe and effective. They review the establishment inspection reports and recalls.
- Office of Communication, Education, and Radiation Programs – responsible for the education of the staff through the Staff College.
- Office of Device Evaluation – responsible for all the pre-market reviews of applications, the pre-market notifications 510(k)s, the pre-market approval PMAs, as well as the investigational device exemptions.
- Office of In-Vitro Diagnostic Device Evaluation and Safety – reviews all the pre-market applications for in vitro diagnostics, as well as all the post-marketing activities associated with in vitro diagnostics.
- Office of Science and Engineering Laboratories – they do independent lab testing, work with standards organizations, universities, and other academia.
- Office of Surveillance and Biometrics – responsible for the post-marketing activities or adverse event reports with regard to medical devices.
Before any medical device can be sold, it must be cleared for use by a trained medical professional at the CDRH.
To do this, the device must be classified based on what type of device it is, and what kind of regulation is required to ensure safety.
- Class I: General Controls – are considered low-risk, generally safe, and may be excluded from the regulatory process all together in some cases.
- Class II: General Controls and Special Controls – These are medium to moderate risk devices.
- Class III: General Controls and Premarket Approval – these are high risk, generally life-supporting, life-sustaining devices.
Let’s talk more about these terms, General Controls, Special Controls, and Premarket Approval.
General Controls require that all finished device manufacturers comply with the FDA’s quality system regulations, manufacturing practices, and are properly labeled.
They also require all manufacturers, user facilities, and importers to report when there’s been an adverse event with a medical device.
This is known as the Medical Device Reporting (MDR).
Special Controls offer some sort of guidance for manufacturing and usage of the product.
For example, a glove manual will give manufacturers specific information about how to prepare a 510(k) application [more on this in just a moment] for a medical glove.
It could also be a mandatory performance standard the product must adhere to. Or it could be a special labeling of some kind.
And last but not least…
510(k) Clearance & Premarket Approval (PMA)
The 510(k) is a marketing clearance process. The purpose is to demonstrate that the device in question is substantially equivalent to another legally-marketed device.
A 510(k) is required anytime a device is introduced for the first time under the applicant’s name, or if they make significant changes that would require new clearance.
But what happens if there isn’t any real equivalent device to compare to? Or what if it’s high risk?
In these cases, the applicant needs to get Pre-Market Approval (PMA).
The purpose of the PMA is to demonstrate to the FDA – through valid scientific evidence – the device is reasonably safe.
Not surprisingly, there are fees the applicant must pay along the way for most of these things.
And like any bureaucratic process, it can move as fast (or as slow) as it wants to.
If the device does get approved, there is then a new set of Post-Market Studies that will need to get done to collect long-term safety and/or effectiveness data.
One of these studies is called post-approval study and is required as a condition of approval for a PMA.
But wait, there’s more…
Section 522 Post-market Surveillance Studies are studies that are mandated by FDA any time after a 510(k) is cleared or a PMA is approved. The criteria for a 522 study for a Class II or Class III device are:
- Failure of the device would be reasonably likely to have a serious adverse health consequence;
- It’s expected to have significant use in pediatric populations;
- It’s intended to be implanted in the body for more than 1 year; or
- It’s intended to be a life-supporting device used outside of the user facility.
Normally, these studies last for up to three years.
So, to bring it all back…
Before investing in a medical device deal, you’ll want to know four main things:
- How is this device classified?
- Has it been granted 510(k) clearance?
- Does it require a Pre-Market Approval?
- What are the Post-Market Studies requirements?
Remember, at the bottom of every Regulation Crowdfunding offering is the investor Q&A forum.
If you don’t understand something or need clarification, ask them!
And if for any reason, you just can’t wrap your head around how this device can be brought to market and effectively commercialized…
That might be a good sign the deal isn’t right for you.
Yours for investing equality,
![]()
Jordan Gillissie – CEO
Equifund